

Associated Faculty
Chen Huang
Shangchao Lin
William Oates
Per Arne Rikvold
Sachin Shanbhag
Jose L. Mendoza-Cortes
Research Example
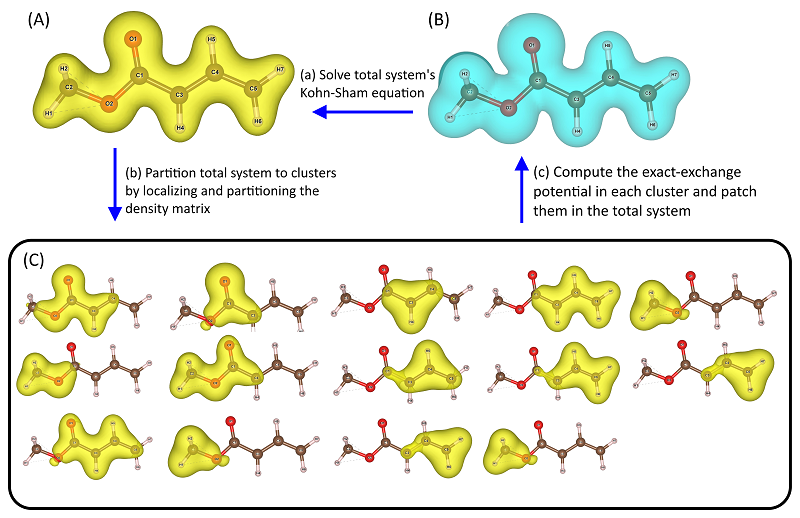
The figure illustrates the basic idea of the exchange-correlation potential patching (XCPP) method developed in Huang’s group. XCPP aims to overcome the length-scale limitation in solving the Schrödinger equation, by constructing accurate exchange-correlation potential in materials. It bridges Kohn-Sham density functional theory and sophisticated correlated wave-function methods. The plots demonstrate how to construct the exact-exchange potential in an ester molecule using XCPP. (A) The electron density of the molecule. (B) The total density is decomposed to cluster densities. The partitioning is done by localizing the canonical molecular orbitals. For instance, the first subplot in (B) is a cluster centering at the carbon atom C1. The cluster contains the O2 and C3 atoms which are the neighbors of C1. (C) The total system’s exact-exchange potential. We compute the exact-exchange potential in each cluster, and project it to the central atom. The projected exact-exchange potentials are patched over the total system to form the total exact-exchange potential which is then used as the input for solving the total system’s Kohn-Sham equation. The loop is then closed. We are currently implementing high-level correlation functionals in XCPP to make it a computational tool with prediction power. We plan to employ XCPP to study the complicated electronic structures of functional materials, such as superconductors and magnetic materials.